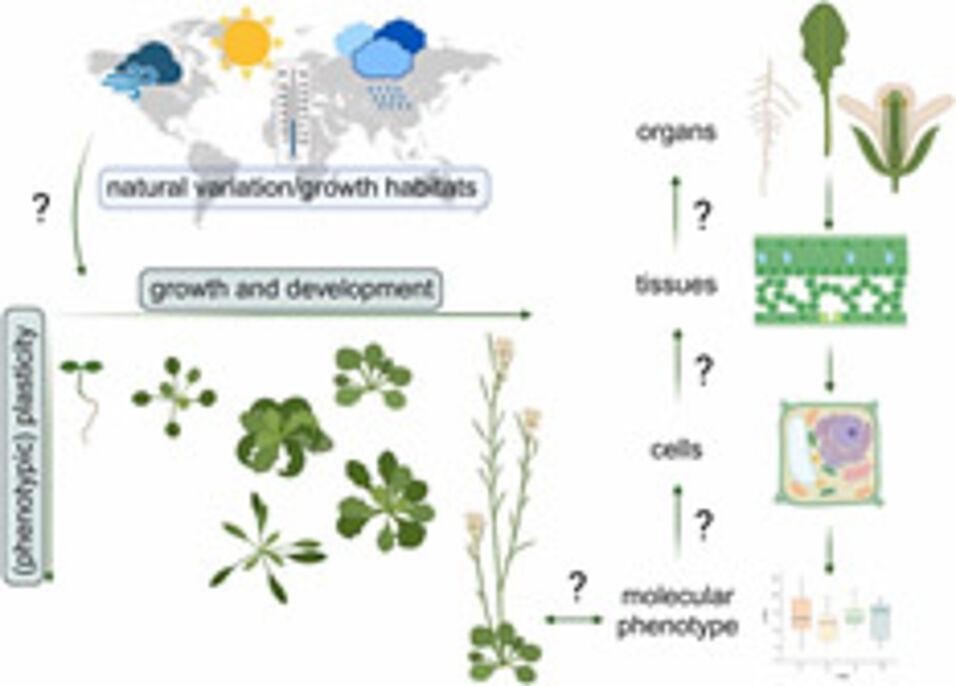
A plant's genome encodes enzymes, transporters and many other proteins which constitute metabolism. Interactions of plants with their environment shape their growth, development and resilience towards adverse conditions.
Although genome sequencing technologies and applications have experienced triumphantly rapid development during the last decades, enabling nowadays a fast and cheap sequencing of full genomes, prediction of metabolic phenotypes from genotype × environment interactions remains, at best, very incomplete. The main reasons are a lack of understanding of how different levels of molecular organisation depend on each other, and how they are constituted and expressed within a setup of growth conditions. Phenotypic plasticity, e.g., of the genetic model plant Arabidopsis thaliana, has provided important insights into plant-environment interactions and the resulting genotype x phenotype relationships.
Here, we summarize previous and current findings about plant development in a changing environment and how this might be shaped and reflected in metabolism and its regulation. We identify current challenges in the study of plant development and metabolic regulation and provide an outlook of how methodological workflows might support the application of findings made in model systems to crops and their cultivation.
Read the full text here: